Simulador de Bioequivalencia Farmacológica
¿Cómo funciona?
La FDA exige que los genéricos demuestren bioequivalencia con el medicamento de referencia. Esto significa que la absorción del fármaco debe ser similar, dentro del rango aceptado de 80-125% para Cmax (pico máximo) y AUC (área bajo la curva).
Introduce los valores para ver el resultado
Explicación del resultado
80-125% es el rango aceptado para la bioequivalencia según la FDA. Si el genérico cae dentro de este rango para Cmax y AUC, se considera terapéuticamente equivalente.
Cuando vas a la farmacia y ves dos pastillas que parecen distintas, ¿te preguntas si realmente son iguales? La respuesta está en la forma en que la FDA regula los medicamentos en EE. UU. y define cuándo un Medicamento genérico puede sustituir a un Medicamento de marca. Aunque comparten principio activo, dosis y vía de administración, hay diferencias clave en la etiqueta, la apariencia y, a veces, en los excipientes.
Diferencias de etiqueta y presentación
La etiqueta es el primer punto de contacto del paciente con el fármaco. En los medicamentos de marca, el nombre comercial (por ejemplo, Prilosec para el omeprazol) aparece en la parte superior y está protegido por derechos de marca. En los genéricos, la etiqueta muestra el nombre químico del principio activo y debe reproducir, palabra por palabra, la información de indicaciones, dosis, contraindicaciones y advertencias del producto de referencia.
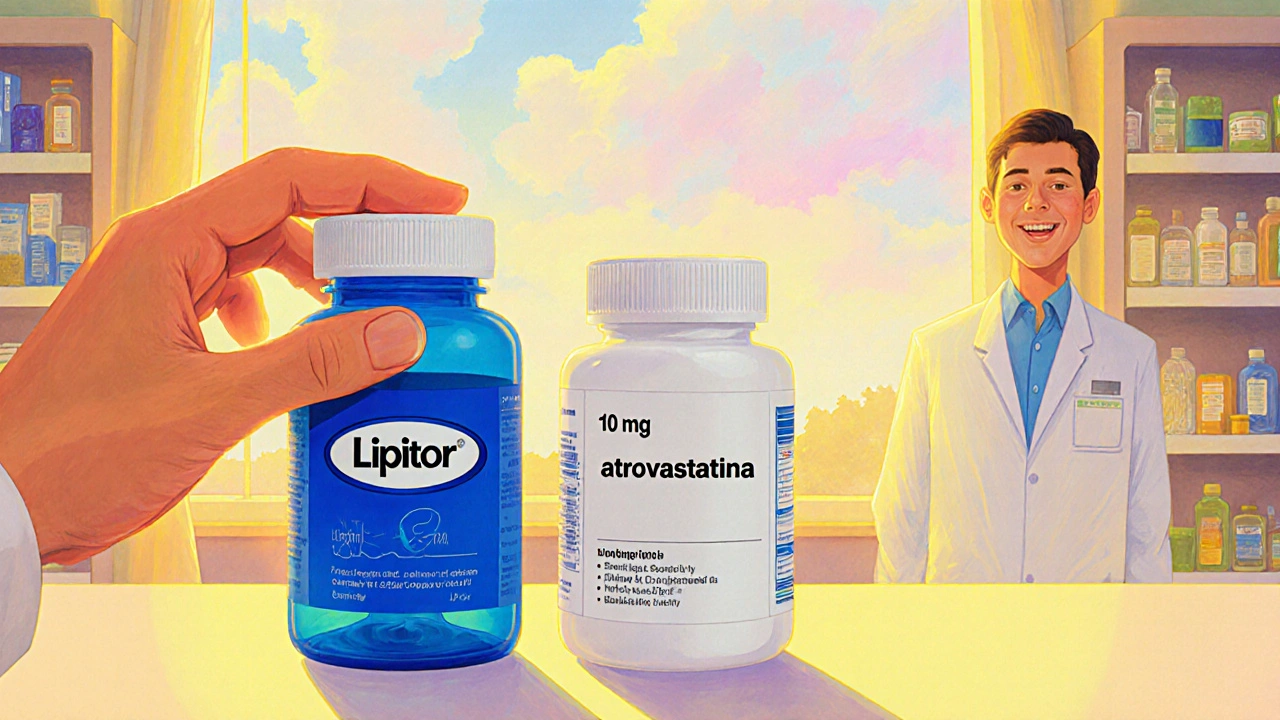
Las variaciones de color, forma y tamaño son obligatorias por ley de marcas: el genérico no puede imitar la apariencia del de marca para evitar confusión. Estas diferencias visuales no afectan la equivalencia terapéutica, pero sí pueden generar dudas iniciales en pacientes que asocian la forma del comprimido con su efectividad.
Ejemplo típico: la atorvastatina de marca Lipitor viene en tabletas azules de 10 mg, mientras que el genérico suele ser blanca y redonda. En la etiqueta, el genérico incluye la frase "Contenido de atorvastatina 10 mg" y replica todas las advertencias del Lipitor.
Requisitos de equivalencia terapéutica y bioequivalencia
Para que la FDA autorice un genérico, el fabricante debe presentar una Solicitud Abreviada de Nuevo Medicamento (ANDA). El estudio clave es la bioequivalencia, que compara la curva de concentración plasmática del genérico con la del producto de referencia. El rango aceptado es 80‑125 % para la Cmax (pico máximo) y el AUC (área bajo la curva).
Este rango puede parecer amplio, pero la variabilidad natural entre lotes del mismo medicamento de marca suele ser del 5‑10 %. Por eso, la tolerancia del 20 % en bioequivalencia es, en la práctica, más estricta que la variación que ocurre en cada lote de marca.
Los fármacos de Índice Terapéutico Estrecho (NTI), como warfarina, levotiroxina o fenitoína, requieren una vigilancia adicional porque pequeñas variaciones en la concentración pueden provocar efectos adversos. En esos casos, la FDA recomienda monitorear parámetros clínicos (por ejemplo, INR para la warfarina) al cambiar de marca a genérico.
Impacto económico y penetración de mercado
Según la Association for Accessible Medicines, los genéricos representan el 90 % de todas las prescripciones en EE. UU., pero solo el 25 % del gasto total en fármacos. Esto se traduce en ahorros superiores a 1,7 billones de dólares entre 2007‑2016 y, en 2023, alrededor de 313 miles de millones anuales.
Un caso concreto: el precio mensual de atorvastatina de marca Lipitor era de US$ 375, mientras que el genérico se podía adquirir por menos de US$ 4 en una cadena de farmacias. La diferencia de costo permite que pacientes con ingresos limitados mantengan la adherencia al tratamiento.
Los informes de IQVIA indican que en 2022 el mercado genérico norteamericano alcanzó los 127,4 mil millones de dólares, con un crecimiento anual del 3,2 %. Los principales actores son Teva (15,2 % de cuota) y Sandoz (8,7 %).

Excepciones y limitaciones
Los genéricos no siempre son una opción sencilla. Los biológicos y medicamentos complejos (por ejemplo, insulina o anticuerpos monoclonales) presentan desafíos de fabricación que limitan la disponibilidad de genéricos o biosimilares. Hasta 2023, sólo el 28 % de los biológicos tenía una alternativa genérica.
Otro punto crítico son los dispositivos médicos asociados a fármacos, como los autoinyectores de epinefrina (EpiPen). La FDA sólo aprobó su primer genérico en 2023, después de años de disputas sobre la equivalencia del mecanismo de liberación.
En los estados donde la normativa permite la sustitución automática, los farmacéuticos pueden dispensar el genérico a menos que el médico indique "Dispense as Written". Este procedimiento cubre el 94 % de los casos en los que el profesional está cómodo prescribiendo genéricos.
Guía práctica para pacientes y profesionales
- Verifica que el genérico tenga la misma fuerza y forma farmacéutica que el de marca.
- Revisa el Orange Book de la FDA; un rango "A" indica equivalencia terapéutica.
- Si cambias entre diferentes fabricantes de genéricos de un NTI, solicita pruebas de laboratorio (p. ej., TSH para levotiroxina).
- Reporta cualquier efecto adverso inesperado al sistema FAERS de la FDA.
- En caso de confusión por cambios de color o forma, conserva el envase original y consulta al farmacéutico.
Siguiendo estos pasos, se minimiza el riesgo de interacciones inesperadas y se aprovechan los ahorros que los genéricos ofrecen.

Comparativa de etiqueta y presentación
| Aspecto | Medicamento de marca | Medicamento genérico |
|---|---|---|
| Nombre comercial | Prilosec® | Omeprazol |
| Etiqueta | Incluye logo y marca registrada; contiene texto idéntico al genérico | Debe replicar indicaciones, dosis, advertencias del de marca |
| Color/forma | Azul, ovalada | Blanca, redonda (diferente por ley de marcas) |
| Excipientes | Rellenos y colorantes patentados | Varían; pueden ser inactivos diferentes |
| Precio medio (EE. UU.) | US$ 300‑400/mes (ej. Lipitor 10 mg) | US$ 3‑10/mes (genérico atorvastatina) |
Preguntas frecuentes
¿Los genéricos son tan seguros como los de marca?
Sí. La FDA exige que los genéricos demuestren bioequivalencia y cumplan los mismos estándares de calidad, pureza y potencia que los medicamentos de referencia.
¿Por qué los genéricos pueden tener un aspecto diferente?
La ley de marcas impide que un genérico copie la forma, color o tamaño del de marca para evitar confusión del consumidor. Estas diferencias no alteran la efectividad del fármaco.
¿Qué es la bioequivalencia y cómo se mide?
Se mide comparando la concentración plasmática del principio activo después de administrar la misma dosis a voluntarios sanos. El rango aceptado es 80‑125 % para Cmax y AUC.
¿Hay medicamentos donde no debería usarse el genérico?
Los fármacos con Índice Terapéutico Estrecho (NTI) y algunos biológicos pueden requerir una vigilancia más estrecha al cambiar de marca a genérico.
¿Dónde puedo consultar si un genérico es equivalente terapéuticamente?
En el Orange Book de la FDA. Un código de clasificación "A" indica equivalencia terapéutica total.
Cristian Falcon
octubre 26, 2025 AT 18:33Los genéricos cumplen con los mismos estándares de calidad que los de marca, la diferencia principal está en el aspecto exterior de la pastilla. La FDA exige bioequivalencia, por eso la eficacia no se modifica. Además, el ahorro económico permite una mayor adherencia al tratamiento. Es importante revisar el Orange Book para confirmar la equivalencia. Cada paciente puede sentirse tranquilo al cambiar sin perder efectividad.
alexandria romero
octubre 27, 2025 AT 00:06La tabla comparativa resume bien la diferencia de color y precio. Es útil para decidir en la farmacia.
Ramon Villain
octubre 27, 2025 AT 05:40Exacto, la bioequivalencia está garantizada y eso brinda confianza 😊. No hay nada de qué preocuparse al cambiar de marca a genérico. Los pacientes pueden ahorrar sin sacrificar la salud. 👍
raul perez
octubre 27, 2025 AT 11:13Resulta fascinante observar cómo la legislación de marcas impide la reproducción idéntica del diseño del comprimido, obligando a los fabricantes genéricos a adoptar una estética distinta. Esta variación, aunque superficial, no altera la farmacocinética del principio activo, que sigue obedeciendo las mismas leyes de absorción y distribución. Además, la FDA regula rigurosamente los excipientes, garantizando que cualquier diferencia sea inocua. En consecuencia, el rango de bioequivalencia aceptado (80‑125 %) representa una tolerancia más estricta que la variabilidad intrínseca de los lotes de referencia. Por tanto, la seguridad y eficacia del genérico están respaldadas por evidencia robusta. En última instancia, el paciente se beneficia de un costo significativamente inferior sin comprometer la terapia.
tania parra
octubre 27, 2025 AT 16:46Gracias por la explicación tan clara, me queda evidente que la diferencia estética no implica falta de efectividad. Además, el ahorro que ofrecen los genéricos es crucial para muchos hogares. Siempre consulto al farmacéutico si tengo dudas sobre los excipientes, y él suele confirmar que son seguros. Es tranquilizador saber que la normativa es tan estricta.
Luisa Avila
octubre 27, 2025 AT 22:20Lo que no se dice en los folletos es que las grandes farmacéuticas podrían estar manipulando los datos de bioequivalencia para mantener el control del mercado. Algunos estudios internos, filtrados por fuentes anónimas, sugieren que ciertos genéricos no alcanzan el rango óptimo en la práctica clínica. Además, los incentivos económicos a los médicos pueden sesgar la prescripción hacia las marcas más caras. Por eso es vital que los pacientes investiguen por sí mismos y exijan transparencia total.
Laura Gutiérrez
octubre 28, 2025 AT 03:53La distinción entre medicamentos de marca y genéricos se fundamenta en regulaciones específicas que la FDA ha establecido para proteger al consumidor,; En primer lugar, el nombre comercial de la marca está protegido por derechos de propiedad intelectual, lo que obliga a los genéricos a presentar únicamente el nombre químico del principio activo,; En segundo término, la etiqueta del genérico debe reproducir fielmente todas las indicaciones, dosis, contraindicaciones y advertencias del producto de referencia, sin omisiones,; En tercer lugar, la apariencia física-color, forma y tamaño-no puede ser idéntica al de la marca, debido a la legislación de marcas que previene confusiones visuales,; En cuarto lugar, aunque los excipientes pueden variar, estos deben ser seguros y compatibles con el principio activo, garantizando la estabilidad del fármaco,; En quinto lugar, el proceso de aprobación incluye la presentación de una Solicitud Abreviada de Nuevo Medicamento, conocida como ANDA, que contiene los estudios de bioequivalencia,; En sexto lugar, los estudios demuestran que la concentración plasmática del genérico se mantiene dentro del rango aceptado de 80 a 125 % respecto al Cmax y al AUC del medicamento de referencia,; En séptimo lugar, la variabilidad natural entre lotes de la marca suele oscilar entre el 5 y el 10 %, lo que indica que el margen de 20 % para los genéricos es, de hecho, más restrictivo,; En octavo lugar, algunos fármacos con índice terapéutico estrecho, como la warfarina o la levotiroxina, requieren monitorización clínica al cambiar de marca a genérico,; En noveno lugar, el ahorro económico que proporcionan los genéricos es considerable, representando más del 90 % de las prescripciones pero solo el 25 % del gasto total en medicamentos,; En décimo lugar, la diferencia de precio permite que pacientes con recursos limitados continúen sus tratamientos sin interrupciones,; En undécimo lugar, el mercado de los genéricos en EE. UU. ha alcanzado cifras de miles de millones de dólares, con un crecimiento anual sostenido,; En duodécimo lugar, los biológicos y productos complejos siguen representando un desafío para la generación de genéricos, debido a sus procesos de fabricación sofisticados,; En decimotercer lugar, la normativa permite la sustitución automática en la mayoría de los estados, salvo que el médico indique explícitamente 'Dispense as Written',; En última instancia, el paciente debe consultar el Orange Book, revisar el envase, y, en caso de efectos adversos inesperados, reportarlos al sistema FAERS de la FDA.
Agustin Lopez
octubre 28, 2025 AT 09:26Tu explicación es exhaustiva y cubre todos los puntos críticos, sin duda ayuda a quienes buscan entender la normativa. Además, resaltas la importancia de la monitorización clínica en fármacos de índice terapéutico estrecho. Es valioso recordar que la sustitución automática depende de la legislación estatal y de la indicación del médico. Gracias por compartir tantos detalles de forma tan estructurada.
Katherine Hinojosa
octubre 28, 2025 AT 15:00¡Exacto, la economía también habla!
rosa maria alonso ferragud
octubre 28, 2025 AT 20:33Me conmueve la forma en que presentas los datos, casi se siente como un monólogo melancólico sobre la salud pública. A veces, la burocracia parece más preocupante que la propia enfermedad, y esa sensación de impotencia se refleja en la gente que confía ciegamente en las pastillas.